联系电话:020-88888888
联系电话:020-88888888
Project Optimus,这次FDA是认真的。
陆续听闻几位业内同仁说起手头的项目,从FDA收到了关于剂量优化的意见。
上周有幸第三次听亚宁博士讲Project Optimus,还是常学常新。
剂量优化近来已经成为肿瘤新药在FDA绕不开的话题,结合公开资料以及FDA发布的指导原则草案,一起看看Optimus如何源起,又会将肿瘤新药研发带往何方。
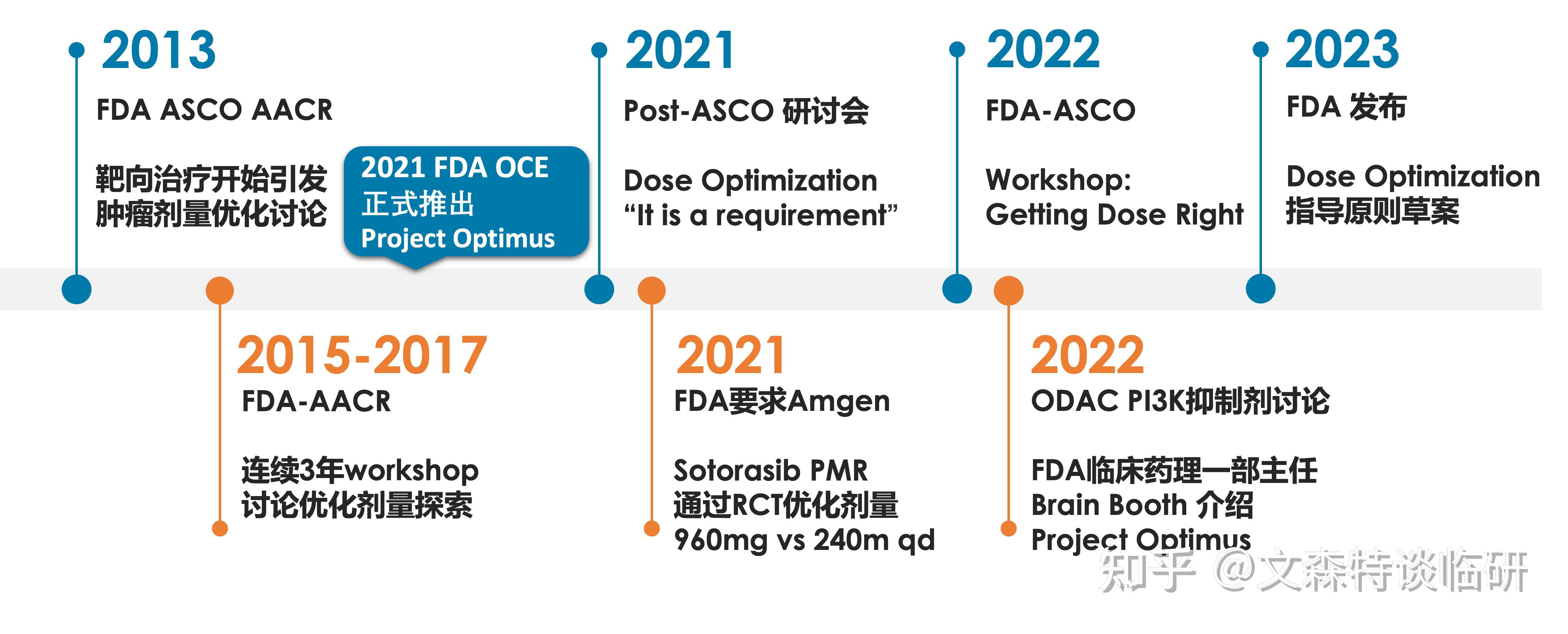

Project Optimus 是FDA肿瘤卓越中心(Oncology Centre of Excellence, OCE)发起的一项旨在改革肿瘤药物开发中剂量优化和剂量选择模式的项目。
Project Optimus 的目标是要推动优化的剂量选择策略,不仅要最大化药物疗效,还要最大限度提高患者的安全性和耐受性。
FDA 鼓励药物开发者在项目早期阶段就与其讨论,综合评估非临床和临床数据,制定包括随机研究在内的剂量探索和剂量优化策略。
一言以蔽之,FDA要改变传统的基于MTD的肿瘤药物剂量选择模式。
"We are going to start making this a requirement.”
-- Richard Pazdur, FDA OCE Director
(1) 剂量效应关系的变化
抗肿瘤药物研发在传统细胞毒化疗时代,常直接选择最大耐受剂量(maximum tolerated dose, MTD)或接近MTD的剂量作为后续开发剂量。
主要原因是缺少精准靶点的药物,呈现陡峭的剂量效应关系。而患者面对危及生命的疾病,也不得不承受更大剂量伴随的毒性。
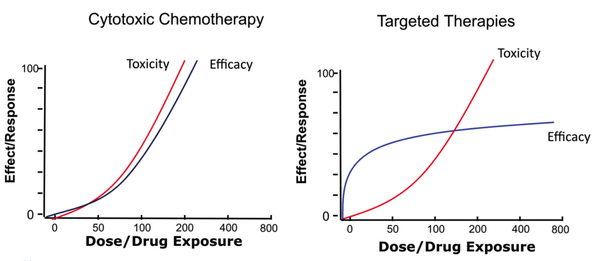
而包括靶向治疗在内的新型疗法,则表现出不同的量效关系。MTD剂量之下的多个剂量,伴随着靶点饱和,疗效并没有随着剂量继续增加,出现“疗效平台”,而脱靶毒性则持续增大。
这个时候,传统3+3探索MTD,直接作为注册研究的剂量就不合理。虽然包括BOIN,mTPI在内的新型剂量爬坡方式可以提高寻找MTD的效率和可靠性,但对于其他剂量仍旧探索较少,剂量选择模式需要进一步优化。
参考前文:剂量爬坡,除了3+3你还需要知道这些
(2) 不合适剂量的问题
不合适的剂量,会影响临床疗效,限制联合及后线治疗,并导致生活质量的降低。
未经优的剂量会影响临床疗效,过高的剂量调整使得患者无法获得最佳的临床获益。剂量未经优化,被FDA认为是限制了Sotorasib和一系列PI3K抑制剂显示生存获益的原因之一。
另一方面,毒性限制了联合治疗以及后线治疗的应用。对于肿瘤后线治疗,经历过不良反应的患者更难耐受,尤其是有叠加毒性的时候。
不仅如此,靶向用药的时间往往更长,低级别但持续的系统毒性,也往往严重影响了生活质量。
如今,剂量优化这个在其他治疗领域的常规要求,通过Project Optimus 也来到了肿瘤治疗。
总体来说,剂量选择模式的转变,将改变早期临床研究,也对临床开发计划和预期造成影响。
Optimus 适用于所有提交FDA药品评价与研究中心( CDER)的治疗性药物和生物制剂。提交生物制品评价与研究中心(CBER)的核药、细胞基因治疗、肿瘤疫苗等除外。
FDA的加速审评计划,包括获得BTD资格的药物也不能豁免。发病率罕见,亦或是同类药物有过剂量研究,也不是免除剂量优化策略的理由。
不仅如此,FDA 还在回溯性审查进行中的研究,如果缺少合理依据,也可能造成临床开发和审查进度的影响。
因此,肿瘤新药都需要在注册临床前充分优化剂量,没有例外。
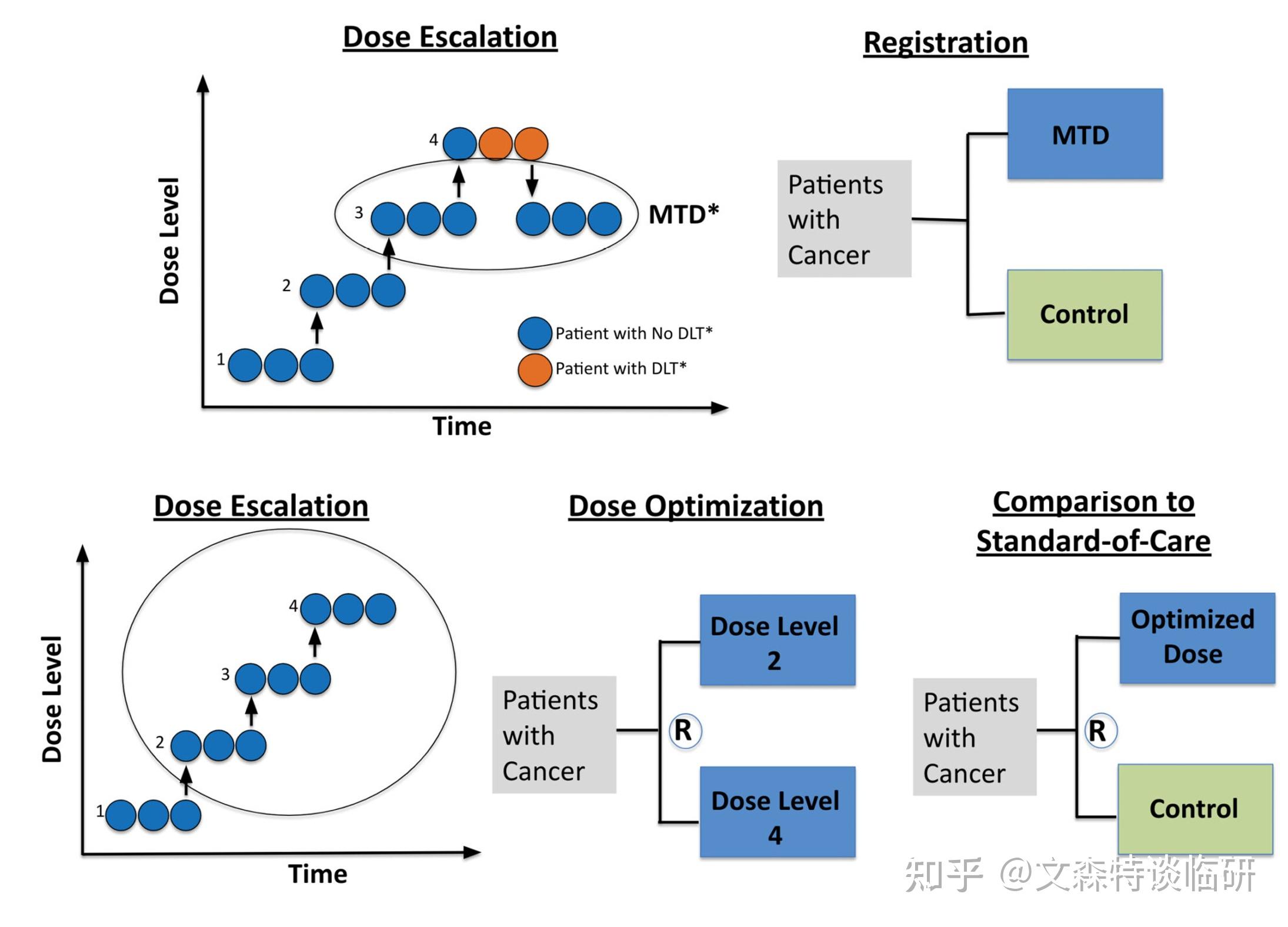
(1) 早期研究及开发计划的变化
剂量探索阶段需要增加患者人数。在剂量爬坡和初始剂量扩展之后,需要识别几个候选剂量或剂量范围,进一步通过随机研究评估不同剂量。
观察期需要延长。在此阶段,相比原有以DLT为主的安全性评估,还需要进一步结合剂量调整、延迟毒性以及可能影响用药的低级别但持续性毒性。
由此将带来临床开发计划的影响。额外的队列研究以及更长的观察期,进入确证性研究会相应推迟。与此同时,在早期探索中优化剂量,试验设计和执行的复杂性会增加,这些也都会带来成本的增加。
(2) 更多的FDA沟通
Optimus意味着,剂量优化策略需要作为开发阶段与FDA沟通确认的重要内容。
比如preIND阶段,需要确认剂量爬坡和早期活性评估的方法;EOP1就拓展评估的剂量达成一致;进入注册研究之前,更需要就研究剂量达成一致。与此同时,优化剂量下的新型试验设计,也需要与FDA充分沟通。
FDA声明,缺少充分剂量选择依据的研究,会使患者暴露于不合理的风险,是明确不可接受的。因此临床研发过程中,需要更多的沟通,减少影响开发进度的风险。
(3) MIDD的应用
Optimus 提示整合临床数据,通过定量药理优化剂量选择的重要性(MIDD),在此不赘述。
参考前文:MIDD | 什么是模型引导的药物研发
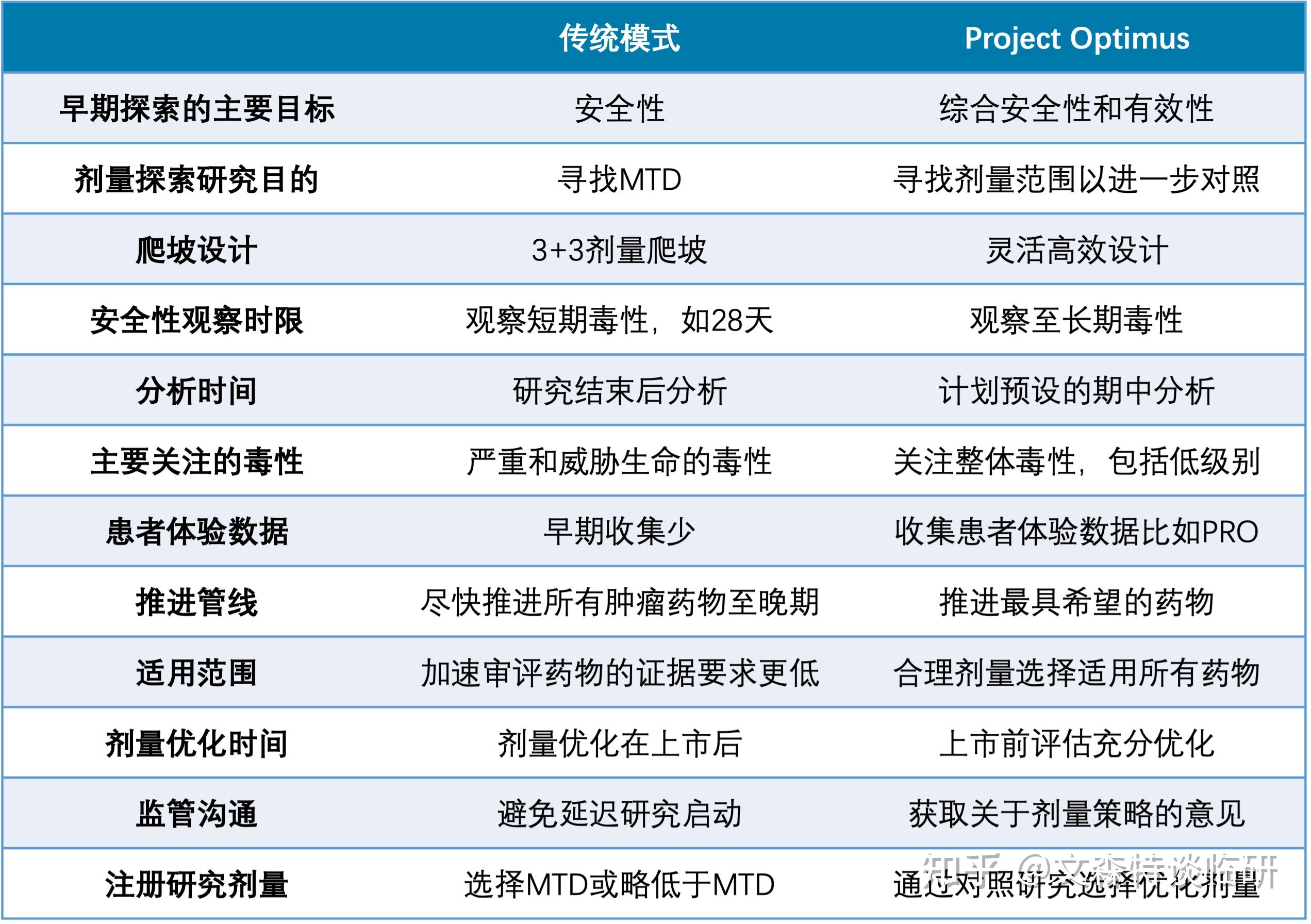
随机平行队列:剂量优化研究尽可能选择随机平行队列,避免序贯研究,以保证接受各剂量组患者的相似性,增加结果的可解释性。
剂量选择:选择至少2个剂量,基于前期非临床和临床信息,对量效关系的初步理解。
最低剂量:选择依据PK/PD分析预计具有活性的最低剂量;最高剂量:安全性允许范围内,用以确定是否在可接受毒性下增加活性;要求两者之间在药代动力学上可区分,且没有重叠PK暴露,比如剂量相差 2-3 倍。
样本量:不需要统计学优效/非劣,但需要足以评估疗效和安全性耐受性,初步描述剂量效应关系。队列样本量并未在指导原则中明确,按照一般解读,比如20-40例。
适应性设计: 可以考虑设置期中分析终止一个或多个剂量组。应用无缝适应性设计可以加快衔接。
参考前文:临床试验的适应性设计:常用类型和考虑因素
评估要点:在不同剂量之间比较用药暴露、完成计划剂量以及AE导致暂停、减量和停药的比例,发生SAE和死亡的比例。
设置安全性监查规则:明确剂量调整或SAE过高时的措施,比如DMC可以建议暂停研究、改变未来给药剂量甚至终止研究。
涵盖低级别不良事件:需要关注影响耐受性的低级别事件,其发生频率和导致的用药影响,事件比如1-2级的腹泻等。
探索不同给药策略:可以依据药物特征,探索提高耐受性的给药策略,比如间歇给药、阶梯式的给药策略(低-高、高-低剂量)。
患者报告结局:重视PRO结果,纳入试验评估,提供对于系统不良事件和功能影响的定量评估。
PK采样和分析计划:剂量探索研究需要包含PK采样和分析计划,足以充分表征不同剂量下的PK特征,并且支持进行安全性和有效性的暴露效应分析。
群体PK 和剂量暴露效应分析:需要和抗肿瘤活性、安全性和耐受性数据一起,来选择剂量进行进一步评估。
特殊人群考虑:入组相对广泛的人群,并且评估特殊人群(考虑体重、年龄、性别、种族、器官功能不全等),并且考虑相关协变量。
生物标志物:考虑药效学标志物以及其他提示临床指标的标志物,比如HIF2α抑制剂患者的EPO水平、FGFR抑制剂患者的血磷酸盐水平,以及ctDNA等可以为剂量优化提供参考。
参考前文:生物标志物的分类和应用
药物制剂:口服药物需要考虑体积和数量带来的药丸负担(pill burden);肠外使用需要考虑浓度和体积。FDA认为工艺难度不是豁免优化剂量的理由。
后续适应症开发:不同疾病分期和肿瘤类型可能有不同的剂量。需要考虑到肿瘤生物学、患者人群以及联合治疗等因素。
比如Enhertu 在FDA获批乳腺癌及肺癌的剂量是 5.4mg/kg Q3W,而在胃癌中则是6.4mg/kg Q3W。
对于新瘤种或新的联治治疗,注册研究之前需要提供充足的剂量选择依据,否则可能会要求额外的剂量探索研究。
今天介绍了FDA Project Optimus,其带来的影响以及需要的改变。
不仅是FDA,各国监管都在关注肿瘤剂量优化策略。
CDE已多次提到给药策略优化,并在法规中强调,包括ADC、双抗以及CLL和AML等技术指导原则。比如ADC临床研发技术指导原则中,提到建议选择多个候选给药方案进行剂量扩展。
以Project Optimus 为代表的剂量优化要求,给传统MTD为基础的肿瘤剂量策略带来变化。适应这种原来被搁置的需求,在研发阶段充分探索优化剂量,才能使患者充分获益。
肿瘤药物剂量,More Isn’t Always Better。
参考文献
CRP聊新药研发与临床试验,关注专栏/公众号:文森特谈临研
 QQ:88888888
QQ:88888888 13899999999
13899999999

 返回顶部
返回顶部
